
Your cells die every day. Don’t worry, your body is protecting itself. In a process known as apoptosis or programmed cell death, cells that are no longer needed commit suicide. Some cells are only required for a short time, they maybe infected by a virus or develop harmful cancerous mutations. Cell death is also an essential part of development from an embryo. For example mouse paws begin as spade-like structures and only form the individual digits as the cells in between die [1]. During apoptosis the cells fragment into smaller apoptotic bodies, and their cell surface is flipped open to display lipid molecules called phosphatidylserines, which act as an “eat me” signal to recruit cells called macrophages to engulf them, before their contents spill out and damage the surrounding tissue. This is a process known as efferocytosis.
However cell death is not always so orderly. Some cells suffer premature death known as necrosis, where they burst open for various reasons such as infection, physical trauma or extreme temperatures. However as the cell’s contents are released into the open, an inflammatory response is triggered, so the macrophages sent to engulf these cells release substances that can damage the surrounding tissue, resulting in a build-up of dead cells. It is this damaging chain of events that often occurs in atherosclerosis; the build-up of fatty plaques which can block arteries or trigger blood clots leading to heart attacks, strokes or tissue death, known as ischaemia. As fatty lipid molecules (primarily LDL or ‘bad’ cholesterol) build up in arteries, they act like damage signals. Macrophages recognise these damage signals as if it is phosphatidylserine, and engulf the lipids to become what is known as a foam cell; a cell full of lipid. A healthy macrophage can repackage the LDL into larger HDL cholesterol, which is released back into the bloodstream to be excreted by the liver. The foam cell can also leave the atherosclerotic plaque to be disposed of via lymphatic vessels, thus shrinking the plaque. However, foam cells can be overwhelmed by engulfing excess cholesterol, increasing harmful inflammatory signals, stress and apoptosis. But all is not lost here. If other macrophages clear the dying foam cells, less harm will be done. The problem is the increased inflammation renders efferocytosis defective, resulting in a process called secondary necrosis. Here apoptotic bodies swell and burst open, as they haven’t been cleared in time. As a result, a large amount of cell debris builds up inside the atherosclerotic plaque, creating what is referred to as a necrotic core. The core is pro-thrombotic when it is exposed to clotting factors in the bloodstream. 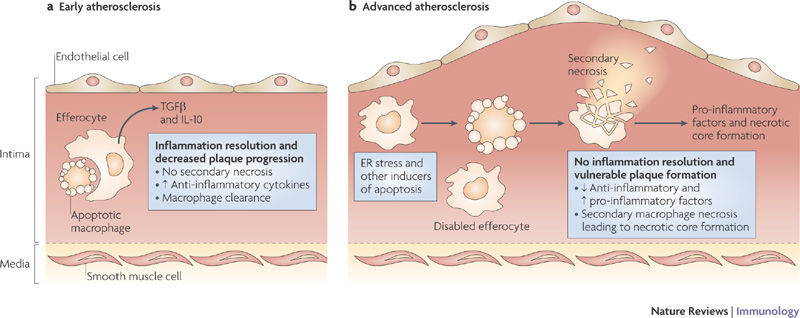
In early atherosclerosis, dying cells are easily cleared. As stress and inflammation increase in advanced plaques, efferocytosis is defective thus secondary necrosis causes necrotic core formation. (Figure 3 from Tabas 2010, Nature Reviews Immunology [2]).
How efferocytosis becomes defective is not entirely understood. During my PhD studies, I was investigating whether inflammation caused by a gene called IRF5 (Interferon Regulatory Factor 5) worsens atherosclerosis as IRF5 is known to generate inflammatory macrophages [3]. My studies not only confirmed this but led my research group down a fascinating path into its effect on efferocytosis, which was recently published in the high impact journal Circulation [4].
We examined arteries from tissues where IRF5 had been deleted versus those with normal levels of IRF5. We already know that deleting IRF5 reduces arthritis and insulin resistance (a precursor to diabetes). Our studies showed that even though body weight fell when IRF5 was deleted, there was much less atherosclerosis in the arteries with smaller necrotic cores, and a reduction in genes associated with inflammation. In fully developed human plaques, IRF5 was present in cells near the necrotic core suggesting it is important for necrotic core formation. This was confirmed when we found cells lacking IRF5 were more resistant to apoptosis and more efficient at efferocytosis than when IRF5 was present. The ability of these cells to perform efferocytosis was dependent on two proteins – Itgb3 and Mfge8, which were both reduced by IRF5. These two proteins form part of a bridge between the macrophage and phosphatidylserine on apoptotic cells. Our findings improved the understanding of necrotic core formation, thus opening the possibility of developing therapies to reduce atherosclerotic plaque formation and limiting the chances of thrombosis. 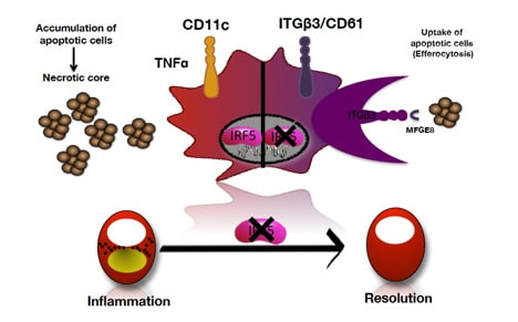
When IRF5 is present (left) dying cells accumulate. But when IRF5 is deleted (right), macrophages can engulf dying cells. (Supplementary figure 8 from Seneviratne et al. 2017, Circulation [4])
Acknowledgments
Many thanks to my PhD supervisors Prof. Claudia Monaco and Prof. Rob Krams, and all members of the Monaco group and the Kennedy Institute of Rheumatology who supported me during my PhD and helped to publish this paper, especially Dr. Jennifer Cole, Michael Goddard, Dr. Andreas Edsfeldt, Dr. Christina Kassiteridi and Inhye Park.
References
[1] Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P, Molecular Biology of the Cell 4th Edition, Garland Science, 2002, https://www.ncbi.nlm.nih.gov/books/NBK26873/ [2] Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nature Reviews Immunology. 2010 ;10(1):36–46. http://www.nature.com/nri/journal/v10/n1/full/nri2675.html [3] Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, Hussell T, Feldmann M, Udalova IA. IRF5 promotes inflammatory macrophage polarization and T(H)1-T(H)17 responses. Nature Immunology 2011 ;12(3):231–8. http://www.nature.com/ni/journal/v12/n3/full/ni.1990.html [4] Seneviratne AN, Edsfeldt AO, Cole JE, Kassiteridi C, Swart M, Park I, Green P, Khoyratty TE, Saliba DG, Goddard ME, Sansom SN, Goncalves I, Krams R, Udalova IA, Monaco C. Interferon Regulatory Factor 5 Controls Necrotic Core Formation in Atherosclerotic Lesions by Impairing Efferocytosis. Circulation. 2017;136:1140-1154 ;http://circ.ahajournals.org/content/early/2017/07/11/CIRCULATIONAHA.117.027844
0 Comments
Your comment will be posted after it is approved.
Leave a Reply. |
AuthorDr. Anusha Seneviratne  This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. 
Categories
All
Archives
March 2020
|
